What is DNA Sequence Alignment?
To compare two or more sequences, it is necessary to align the conserved and unconserved residues across all the sequences (identification of locations of insertions and deletions that have occurred since the divergence of a common ancestor). These residues form a pattern from which the relationship between sequences can be determined with phylogenetic programs. When the sequences are aligned, it is possible to identify locations of insertions or deletions since their divergence from their common ancestor. There are three possibilities :
- The bases match : this means that there is no change since their divergence.
- The bases mismatch : this means that there is a substitution since their divergence.
- There is a base in one sequence, no base in the other : there is an insertion or a deletion since their divergence.
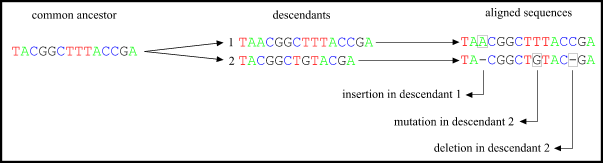 Figure: The comparison of sequences. A good alignment is important for the next step : the construction of phylogenetic trees. The alignment will affect the distances between 2 different species and this will influence the inferred phylogeny. There are several programs available on the net for aligning sequences. These are all based on different mathematical models to compare two or more sequences with the most optimal score for matching bases with a minimum number of gaps inserted (because you can insert a huge amount of gaps, so every base will match an other).
Figure: The comparison of sequences. A good alignment is important for the next step : the construction of phylogenetic trees. The alignment will affect the distances between 2 different species and this will influence the inferred phylogeny. There are several programs available on the net for aligning sequences. These are all based on different mathematical models to compare two or more sequences with the most optimal score for matching bases with a minimum number of gaps inserted (because you can insert a huge amount of gaps, so every base will match an other). Example : two sequences :TCAGACGATTG
TCGGAGCTG
How can we get the best alignment ? There are several possibilities : 1. Reduce the number of mismatches :TCAG-ACG-ATTG2. Reduce the number of gaps :
|| | | | | | 0 mismatches 7 matches 6 gaps
TC-GGA-GC-T-GTCAGACGATTG3. Reduce neither the number of gaps nor the number of mismatches :
|| || 5 mismatches 4 matches 2 gaps
TCGGAGCTG--TCAG-ACGATTG4. Same as 3. but one base (or gap) moved :
|| | | | | 2 mismatches 6 matches 4 gaps
TC-GGA-GCTG-TCAG-ACGATTGWhich of these is now the best alignment ?? There are several alignment algorithms to choose the best alignment. Let's use a simple one in this example :
|| | | | | | 1 mismatch 7 matches 4 gaps
TC-GGA-GCT-G
D = y + sum(wkzk)
with :
D = distance
y : number of mismatches
w : penalty for gaps of length k
z : number of gaps of length k
Take gap penalty for gap length 1 = 2
Take gap penalty for gap length 2 = 6 (short gaps occur more frequent than long gaps)
in 1. : 0 + {(2 x 6) + (6 x 0)} = 12
in 2. : 5 + {(2 x 0) + (6 x 1)} = 11
in 3. : 2 + {(2 x 4) + (6 x 0)} = 10
in 4. : 1 + {(2 x 4) + (6 x 0)} = 9
We choose alignment 4 because it has the minimum distance.
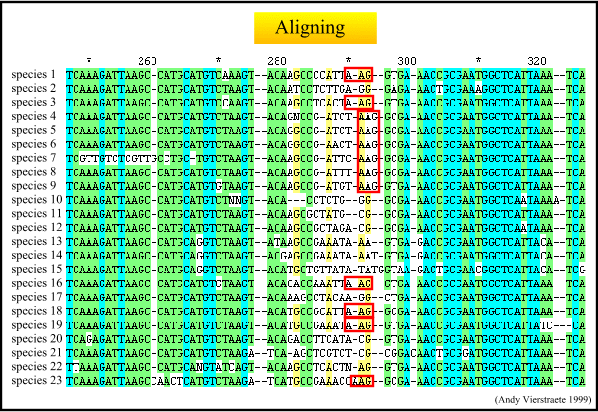 Figure: The alignment of sequences. This is done with Clustalw 1.74, and as you can see, the more variable areas are not optimally aligned (indicated with red boxes). Therefore it is mostly necessary to improve the alignment by hand. In this case, it is obvious to improve the alignment, but in other cases it could be more difficult to make improvements.
Figure: The alignment of sequences. This is done with Clustalw 1.74, and as you can see, the more variable areas are not optimally aligned (indicated with red boxes). Therefore it is mostly necessary to improve the alignment by hand. In this case, it is obvious to improve the alignment, but in other cases it could be more difficult to make improvements.
No comments:
Post a Comment