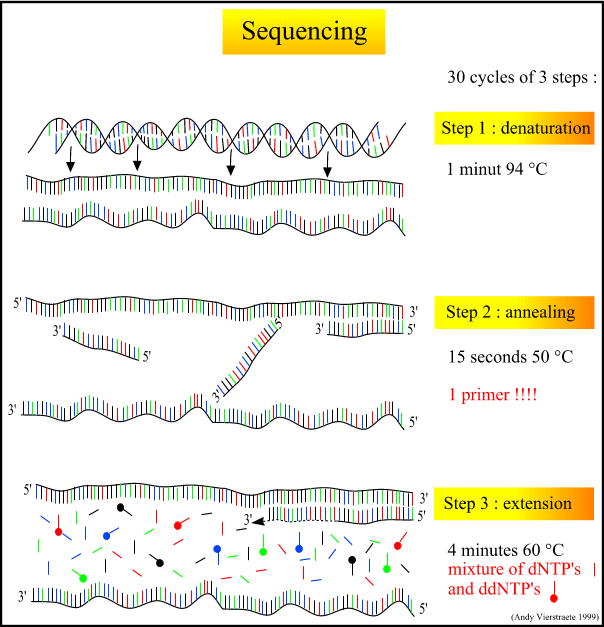
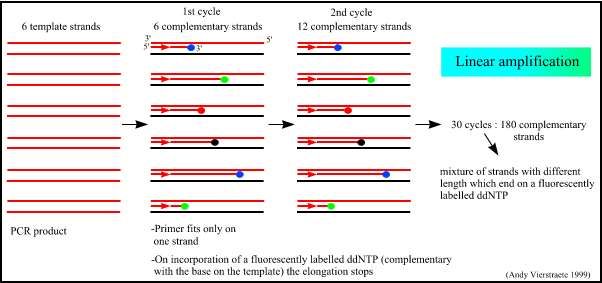
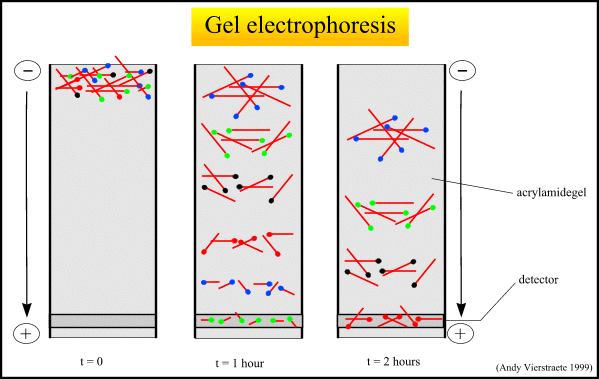
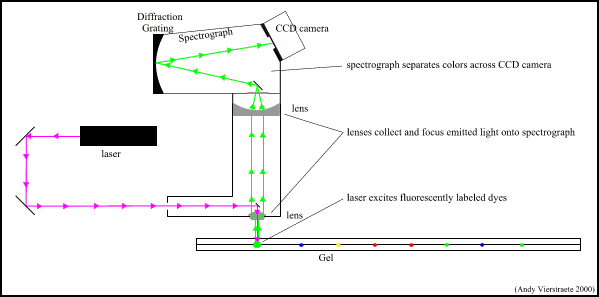
What is DNA Fingerprinting
What is DNA Fingerprinting?
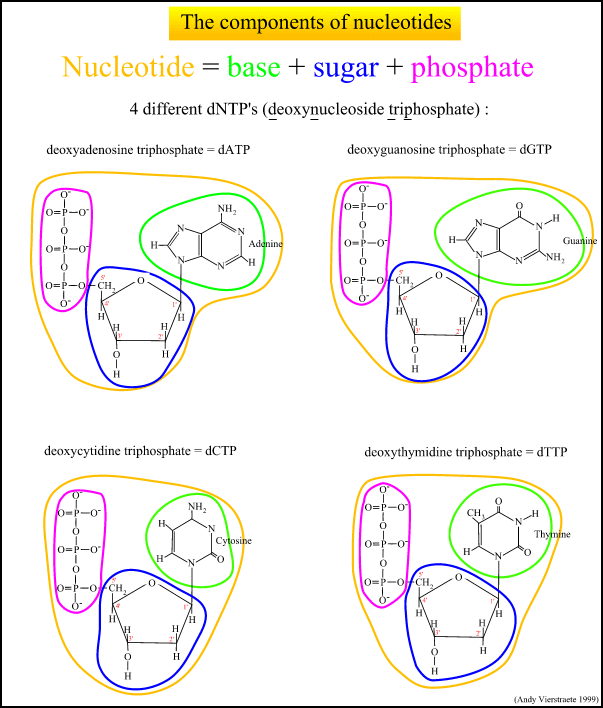
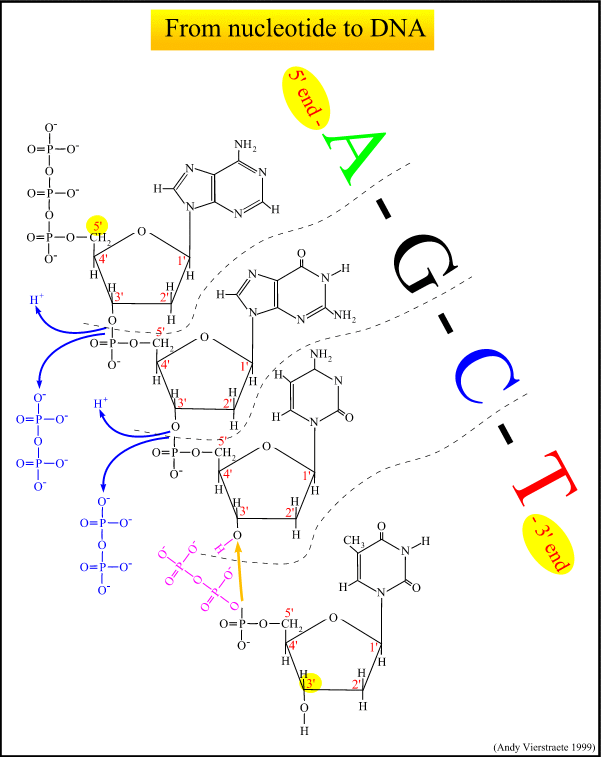
The chemical structure of everyone's DNA is the same. The only difference between people (or any animal) is the order of the base pairs. There are so many millions of base pairs in each person's DNA that every person has a different sequence.
Using these sequences, every person could be identified solely by the sequence of their base pairs. However, because there are so many millions of base pairs, the task would be very time-consuming. Instead, scientists are able to use a shorter method, because of repeating patterns in DNA.
These patterns do not, however, give an individual "fingerprint," but they are able to determine whether two DNA samples are from the same person, related people, or non-related people. Scientists use a small number of sequences of DNA that are known to vary among individuals a great deal, and analyze those to get a certain probability of a match.
How is DNA Fingerprinting Done?
1. Performing Sourthen BlotThe Southern Blot is one way to analyze the genetic patterns which appear in a person's DNA. Performing a Southern Blot involves:
1. Isolating the DNA in question from the rest of the cellular material in the nucleus. This can be done either chemically, by using a detergent to wash the extra material from the DNA,or mechanically, by applying a large amount of pressure in order to "squeeze out" the DNA.
2. Cutting the DNA into several pieces of different sizes. This is done using one or more restriction enzymes.
3. Sorting the DNA pieces by size. The process by which the size separation, "size fractionation," is done is called gel electrophoresis. The DNA is poured into a gel, such as agarose, and an electrical charge is applied to the gel, with the positive charge at the bottom and the negative charge at the top. Because DNA has a slightly negative charge, the pieces of DNA will be attracted towards the bottom of the gel; the smaller pieces, however, will be able to move more quickly and thus further towards the bottom than the larger pieces. The different-sized pieces of DNA will therefore be separated by size, with the smaller pieces towards the bottom and the larger pieces towards the top.
4. Denaturing the DNA, so that all of the DNA is rendered single-stranded. This can be done either by heating or chemically treating the DNA in the gel.
5. Blotting the DNA. The gel with the size-fractionated DNA is applied to a sheet of nitrocellulose paper, and then baked to permanently attach the DNA to the sheet. The Southern Blot is now ready to be analyzed.
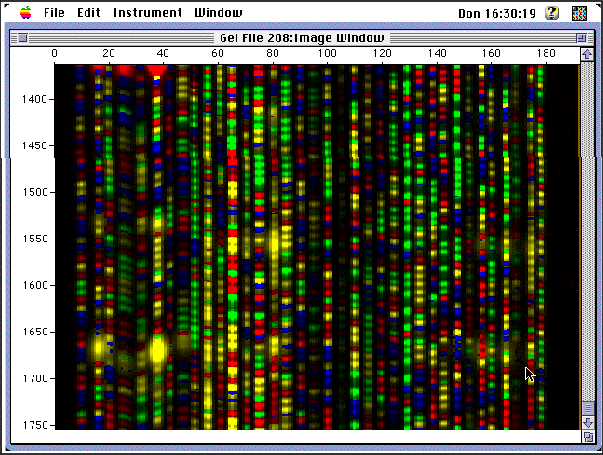
In order to analyze a Southern Blot, a radioactive genetic probe is used in a hybridization reaction with the DNA in question (see next topics for more information). If an X-ray is taken of the Southern Blot after a radioactive probe has been allowed to bond with the denatured DNA on the paper, only the areas where the radioactive probe binds [red] will show up on the film. This allows researchers to identify, in a particular person's DNA, the occurrence and frequency of the particular genetic pattern contained in the probe.

1. Obtain some DNA polymerase [pink]. Put the DNA to be made radioactive (radiolabeled) into a tube.

2. Introduce nicks, or horizontal breaks along a strand, into the DNA you want to radiolabel. At the same time, add individual nucleotides to the nicked DNA, one of which, *C [light blue], is radioactive.

3. Add the DNA polymerase [pink] to the tube with the nicked DNA and the individual nucleotides. The DNA polymerase will become immediately attracted to the nicks in the DNA and attempt to repair the DNA, starting from the 5' end and moving toward the 3' end.

4. The DNA polymerase [pink] begins repairing the nicked DNA. It destroys all the existing bonds in front of it and places the new nucleotides, gathered from the individual nucleotides mixed in the tube, behind it. Whenever a G base is read in the lower strand, a radioactive *C [light blue] base is placed in the new strand. In this fashion, the nicked strand, as it is repaired by the DNA polymerase, is made radioactive by the inclusion of radioactive *C bases.

5. The nicked DNA is then heated, splitting the two strands of DNA apart. This creates single-stranded radioactive and non-radioactive pieces. The radioactive DNA, now called a probe [light blue], is ready for use.

1. Hybridization is the coming together, or binding, of two genetic sequences. The binding occurs because of the hydrogen bonds [pink] between base pairs. Between a A base and a T base, there are two hydrogen bonds; between a C base and a G base, there are three hydrogen bonds.

2. When making use of hybridization in the laboratory, DNA must first be denatured, usually by using heat or chemicals. Denaturing is a process by which the hydrogen bonds of the original double-stranded DNA are broken, leaving a single strand of DNA whose bases are available for hydrogen bonding.

3. Once the DNA has been denatured, a single-stranded radioactive probe [light blue] can be used to see if the denatured DNA contains a sequence similar to that on the probe. The denatured DNA is put into a plastic bag along with the probe and some saline liquid; the bag is then shaken to allow sloshing. If the probe finds a fit, it will bind to the DNA.

4. The fit of the probe to the DNA does not have to be exact. Sequences of varying homology can stick to the DNA even if the fit is poor; the poorer the fit, the fewer the hydrogen bonds between the probe [light blue] and the denatured DNA. The ability of low-homology probes to still bind to DNA can be manipulated through varying the temperature of the hybridization reaction environment, or by varying the amount of salt in the sloshing mixture.

4. VNTRs
Every strand of DNA has pieces that contain genetic information which informs an organism's development (exons) and pieces that, apparently, supply no relevant genetic information at all (introns). Although the introns may seem useless, it has been found that they contain repeated sequences of base pairs. These sequences, called Variable Number Tandem Repeats (VNTRs), can contain anywhere from twenty to one hundred base pairs.
Every human being has some VNTRs. To determine if a person has a particular VNTR, a Southern Blot is performed, and then the Southern Blot is probed, through a hybridization reaction, with a radioactive version of the VNTR in question. The pattern which results from this process is what is often referred to as a DNA fingerprint.
A given person's VNTRs come from the genetic information donated by his or her parents; he or she could have VNTRs inherited from his or her mother or father, or a combination, but never a VNTR either of his or her parents do not have. Shown below are the VNTR patterns for Mrs. Nguyen [blue], Mr. Nguyen [yellow], and their four children: D1 (the Nguyens' biological daughter), D2 (Mr. Nguyen's step-daughter, child of Mrs. Nguyen and her former husband [red]), S1 (the Nguyens' biological son), and S2 (the Nguyens' adopted son, not biologically related [his parents are light and dark green]).

Because VNTR patterns are inherited genetically, a given person's VNTR pattern is more or less unique. The more VNTR probes used to analyze a person's VNTR pattern, the more distinctive and individualized that pattern, or DNA fingerprint, will be.
Pratical Applications of DNA Fingerprinting
1. Paternity and Maternity
Because a person inherits his or her VNTRs from his or her parents, VNTR patterns can be used to establish paternity and maternity. The patterns are so specific that a parental VNTR pattern can be reconstructed even if only the children's VNTR patterns are known (the more children produced, the more reliable the reconstruction). Parent-child VNTR pattern analysis has been used to solve standard father-identification cases as well as more complicated cases of confirming legal nationality and, in instances of adoption, biological parenthood.
2. Criminal Identification and Forensics
DNA isolated from blood, hair, skin cells, or other genetic evidence left at the scene of a crime can be compared, through VNTR patterns, with the DNA of a criminal suspect to determine guilt or innocence. VNTR patterns are also useful in establishing the identity of a homicide victim, either from DNA found as evidence or from the body itself.
3. Personal Identification
The notion of using DNA fingerprints as a sort of genetic bar code to identify individuals has been discussed, but this is not likely to happen anytime in the foreseeable future. The technology required to isolate, keep on file, and then analyze millions of very specified VNTR patterns is both expensive and impractical. Social security numbers, picture ID, and other more mundane methods are much more likely to remain the prevalent ways to establish personal identification.
Problems with DNA Fingerprinting
Like nearly everything else in the scientific world, nothing about DNA fingerprinting is 100% assured. The term DNA fingerprint is, in one sense, a misnomer: it implies that, like a fingerprint, the VNTR pattern for a given person is utterly and completely unique to that person. Actually, all that a VNTR pattern can do is present a probability that the person in question is indeed the person to whom the VNTR pattern (of the child, the criminal evidence, or whatever else) belongs. Given, that probability might be 1 in 20 billion, which would indicate that the person can be reasonably matched with the DNA fingerprint; then again, that probability might only be 1 in 20, leaving a large amount of doubt regarding the specific identity of the VNTR pattern's owner.
1. Generating a High Probability
The probability of a DNA fingerprint belonging to a specific person needs to be reasonably high--especially in criminal cases, where the association helps establish a suspect's guilt or innocence. Using certain rare VNTRs or combinations of VNTRs to create the VNTR pattern increases the probability that the two DNA samples do indeed match (as opposed to look alike, but not actually come from the same person) or correlate (in the case of parents and children).
2. Problems with Determining Probability
A. Population Genetics
VNTRs, because they are results of genetic inheritance, are not distributed evenly across all of human population. A given VNTR cannot, therefore, have a stable probability of occurrence; it will vary depending on an individual's genetic background. The difference in probabilities is particularly visible across racial lines. Some VNTRs that occur very frequently among Hispanics will occur very rarely among Caucasians or African-Americans. Currently, not enough is known about the VNTR frequency distributions among ethnic groups to determine accurate probabilities for individuals within those groups; the heterogeneous genetic composition of interracial individuals, who are growing in number, presents an entirely new set of questions. Further experimentation in this area, known as population genetics, has been surrounded with and hindered by controversy, because the idea of identifying people through genetic anomalies along racial lines comes alarmingly close to the eugenics and ethnic purification movements of the recent past, and, some argue, could provide a scientific basis for racial discrimination.
B. Technical Difficulties
Errors in the hybridization and probing process must also be figured into the probability, and often the idea of error is simply not acceptable. Most people will agree that an innocent person should not be sent to jail, a guilty person allowed to walk free, or a biological mother denied her legal right to custody of her children, simply because a lab technician did not conduct an experiment accurately. When the DNA sample available is minuscule, this is an important consideration, because there is not much room for error, especially if the analysis of the DNA sample involves amplification of the sample (creating a much larger sample of genetically identical DNA from what little material is available), because if the wrong DNA is amplified (i.e. a skin cell from the lab technician) the consequences can be profoundly detrimental. Until recently, the standards for determining DNA fingerprinting matches, and for laboratory security and accuracy which would minimize error, were neither stringent nor universally codified, causing a great deal of public outcry.